On March 22, 2020, the U.S. Food and Drug Administration (“FDA”) issued guidance, for immediate implementation, that aims to increase the availability of ventilators and other respiratory devices needed to address the COVID-19 pandemic. While FDA urges health care facilities to use, wherever possible, FDA-cleared standard full-featured ventilators to treat COVID-19 patients (as well as other patients requiring ventilatory support), FDA will allow a more flexible approach to modifications to these devices to help boost manufacturing capacity and supply. FDA also took the opportunity to lay out guidelines that encourage submission of Emergency Use Authorization (“EUA”) applications for devices not marketed in the United States, continuing an unprecedented Agency response to the pandemic.
Guidance Scope
Specifically, FDA will allow manufacturers of certain FDA-cleared ventilator/respiratory devices (as detailed in the table below) to make modifications to the indications, claims, functionality, or to the hardware, software, or materials of the device without making a new 510(k) submission to FDA, so long as the modification will not create undue risk in light of the public health emergency. Such changes, which would normally require a new 510(k), could include a significant change or modification in design, material, chemical composition, energy source, or manufacturing process.
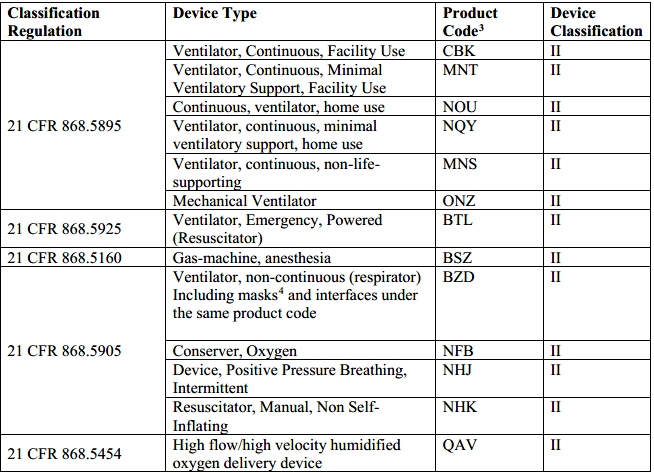
Examples of Acceptable Modifications Without a New 510(k)
Specific examples of device modifications that FDA would not consider to create undue risk are provided below, by category.
Modifications to FDA-Cleared Indications, Claims, or Functionality
- The use of powered emergency ventilators and anesthesia gas machines for patients needing mechanical ventilations;
- The use of ventilators outside their cleared environment of use (for example, use of a ventilator in a health care facility when it is only cleared for use at home or during transport);
- The use of devices indicated for sleep apnea (including the noncontinuous ventilators delivering continuous positive airway pressure (“CPAP”) or bi-level positive airway pressure), provided that appropriate design mitigations are in place to minimize aerosolization; and
- The use of oxygen concentrators for primacy supply when medically necessary and clinically appropriate.
Modifications to Hardware, Software and Material Changes to FDA-Cleared Ventilators and Anesthesia Gas Machines
- Modifications to motors, batteries, or other electrical components;
- Material changes to components in the gas pathway or with other patient tissue contact;
- Introduction of filtration to minimize aerosolization;
- Software modification intended to modify the ventilation parameters;
- Software modification implementing algorithms for oxygen titration where the algorithms/devices are the subject of an FDA-approved Investigational Device Exemption (“IDE”); and
- Hardware and/or software modifications implementing the capability for remote monitoring and remote adjustment of ventilator parameters (e.g., adding wireless and/or Bluetooth capability; also, for such changes, FDA recommends that manufacturers follow FDA guidance regarding cybersecurity).
Use of Ventilator and Anesthesia Gas Machine Breaching Circuit Devices Beyond Their Indicated Shelf Life and Duration of Use
- Modifications in the indicated shelf life and duration of use of the constituent parts of the breathing circuit (e.g., tubing, filters, and humidifiers) used for treating individual patients where devices are used according to healthcare institutional protocols or useful life is limited to the occurrence of malfunction or visible soiling.
Other Considerations When Making Device Modifications
The guidance also provides certain recommendations for manufacturers pursuing the above changes without prior submission of a new 510(k). For example, with respect to changes made to device hardware, software, materials or duration of use, FDA recommends that manufacturers follow FDA recognized standards for the specific device type. Manufacturers must also document changes to the device in the device master record and change control records.
With regard to labeling, FDA recommends that manufacturers use labeling to enhance user understanding of device modifications, including:
- A clear description of the device’s new indications, claims, or functions, and information on the device’s performance and potential risks;
- Adequate instructions for use for the intended user and indicated environment(s) of use. The labeling highlighting the difference in design compared to the unmodified, FDA-cleared version of the device, along with instructions for mitigating any known risks associated with these differences; and
- A distinction delineating FDA-cleared indications and claims from those that are not FDA-cleared. In addition, FDA recommends the labeling include a general statement about changes that have not been cleared by the FDA.
FDA’s Intended Approach for Emergency Use Authorizations (EUAs) for Ventilatory Support Devices
Finally, the guidance notes FDA’s interest in interacting with manufacturers of ventilatory support devices that are not currently marketed in the U.S. FDA asks such manufacturers to send FDA certain delineated information to help the Agency determine whether an EUA can be issued. Once the information is provided, FDA may notify the manufacturer that it does not intend to object to the distribution and use of the device while the manufacturer prepares, and FDA reviews, the EUA request.
FDA is also interested in working with manufacturers without medical device manufacturing experience that may have the capability to help increase the supply of these devices, and encourages such manufacturers to reach out to the Agency describing their proposed approach.
Finally, we note that the U.S. Department of Health and Human Services (“HHS”), the umbrella agency that houses FDA, is also looking for ways to ramp up ventilator production, and recently issued an information request to identify manufacturers that can rapidly produce at least 5,000, and up to 100,000, ventilators (responses were due yesterday, March 23).
Blog Editors
Authors
 Associate
Associate Member of the Firm
Member of the Firm